![[IN]Genios](http://images.squarespace-cdn.com/content/v1/51c861c1e4b0fb70e38c0a8a/48d2f465-eaf4-4dbc-a7ce-9e75312d5b47/logo+final+%28blanco+y+rojo%29+crop.png?format=1500w)
Statistical methods for tumor classification in breast cancer: Supervised and unsupervised approaches
Tatiana M. Vizcarrondo Córdova
Departamento de Estadística y Sistemas Computadorizados de Información
Facultad de Administración de Empresas, UPR RP
María A. Nieves Rivera
Departamento de Matemáticas
Facultad de Ciencias Naturales, UPR RP
Recibido: 19/09/2024; Revisado: 29/10/2024; Aceptado: 04/11/2024
Resumen
Este estudio integra métodos de clasificación de tumores mamarios, combinando enfoques supervisados y no supervisados para mejorar la precisión diagnóstica. Se analizan diez características celulares clave para desarrollar modelos óptimos. Los resultados demuestran una precisión de diagnóstico superior al 97% en su mayoría, prometiendo mejoras significativas en la identificación de tumores benignos y malignos. Estos hallazgos resaltan la efectividad del enfoque propuesto y su relevancia en el diagnóstico preciso del cáncer de mama, instando a continuar mejorando estas técnicas para avanzar en la atención médica de esta enfermedad.
Palabras claves: clasificación, modelo, cáncer, tumor, diagnóstico, datos
Abstract
This study integrates breast tumor classification methods, combining supervised and unsupervised approaches to improve diagnostic accuracy. Ten key cellular features are analyzed to develop optimal models. The results demonstrate a diagnostic accuracy of over 97% in most cases, promising significant improvements in identifying benign and malignant tumors. These findings highlight the effectiveness of the proposed approach and its relevance in the precise diagnosis of breast cancer, encouraging further refinement of these techniques to advance medical care for this disease.
Keywords: classification, model, cancer, tumor, diagnosis, data
Introducción
Este proyecto de investigación tiene como objetivo crear el mejor método de clasificación posible, utilizando tanto técnicas supervisadas como no supervisadas, para distinguir entre tumores benignos y malignos en pacientes con cáncer de mama. Esta tarea es fundamental para mejorar la precisión y la eficacia de los diagnósticos médicos en esta área crucial de la salud. Al combinar métodos supervisados (que utilizan datos etiquetados para entrenar algoritmos de clasificación) con técnicas no supervisadas (que identifican patrones y estructuras ocultas en los datos), buscamos obtener un enfoque completo para la clasificación de tumores mamarios. Esto implica la exploración detallada de las características nucleares y la aplicación de análisis estadísticos avanzados para lograr una clasificación precisa y confiable. El desarrollo de este método de clasificación óptimo no solo tendrá aplicaciones directas en el diagnóstico clínico del cáncer de mama, sino que también sentará las bases para futuras investigaciones en el campo de la oncología y la biomedicina. La capacidad de identificar y distinguir con precisión entre tumores benignos y malignos es crucial para determinar los tratamientos adecuados y mejorar los resultados médicos para las pacientes.
Estuvimos utilizando una base de datos que describe múltiples propiedades de las células cancerígenas. Este conjunto de datos fue adquirido del repositorio de aprendizaje automático de la UC Irvine, una institución destacada en investigación dentro de la red de la Universidad de California, la base de datos consta de 569 observaciones y 32 variables. La información recopilada consiste en características nucleares evaluadas visualmente a partir de aspirados con aguja fina (PAAF) obtenidos de las mamas de pacientes, bajo la supervisión del Dr. William H. Wolberg. Estas características se presentan como variables en nuestra base de datos, cada una identificada por un número único (ID number) junto con el diagnóstico de la muestra como maligna (M) o benigna (B). Entre las características evaluadas se encuentran medidas clave como el radio (distancia promedio del centro a los puntos en el perímetro), textura (desviación estándar de los valores de escala de grises), perímetro, área, suavidad (variación local en longitudes de radio), compacidad (perímetro^2 / área - 1.0), concavidad (gravedad de porciones cóncavas del contorno), puntos cóncavos (número de porciones cóncavas del contorno), simetría y dimensión fractal ("aproximación de la línea costera" - 1). Estas características nos brindan una visión detallada de las propiedades físicas y estructurales de las células mamarias estudiadas, siendo fundamentales para la identificación y el diagnóstico preciso de la presencia de cáncer de mama en las muestras analizadas. El análisis de estas nos puede brindar información valiosa para nuestros modelos de clasificación, contribuyendo así a la investigación en este campo crucial de la salud.
Metodología
En la primera parte del proyecto, nos enfocamos en preparar y limpiar la base de datos para que sea adecuada para el análisis y modelado. Este proceso implica varias etapas para asegurar la integridad y la calidad de los datos. Inicialmente, exportamos nuestra base de datos y procedemos a renombrar las variables utilizando la función “colnames” para que sean más descriptivas y manejables. Luego, verificamos la estructura de la base de datos con la función “skimr::skim”, la cual “nos proporciona un enfoque sencillo para las estadísticas descriptivas que se ajustan al “principio de menor sorpresa”, mostrando estadísticas resumidas que el usuario puede revisar rápidamente para comprender sus datos” (Waring, 2022). Identificamos las variables "Diagnosis" y "ID" y las convertimos a factores, ya que "Diagnosis" es una variable categórica (benigno o maligno) y "ID" es un identificador único para cada muestra. A partir de esta revisión, confirmamos que la base de datos no contiene valores faltantes ni duplicados, lo que es esencial para mantener la precisión de nuestro análisis. Además, determinamos la necesidad de estandarizar las variables numéricas para mejorar la eficacia de los modelos de clasificación que se crearán posteriormente. Para familiarizarnos mejor con la estructura de los datos, creamos varios diagramas exploratorios. Utilizamos la función “ggpairs” de la librería GGally para generar una gráfica que muestra correlaciones gráficas y numéricas entre las variables, así como sus distribuciones individuales. Esta función “proporciona dos comparaciones diferentes de cada par de columnas y muestra ya sea la densidad o el conteo de la respectiva variable a lo largo de la diagonal” (Schloerke, 2015). Esto lo hicimos solo a las variables que representaban la media de alguna de las características en la base de datos. Este análisis visual nos ayuda a identificar relaciones significativas entre las características y a entender mejor la variabilidad dentro de los datos. También, elaboramos un diagrama de barras para visualizar la distribución de los tumores benignos y malignos en la base de datos, lo que nos proporciona una visión clara de la proporción de cada tipo de tumor.
Con nuestra base de datos preparada, pasamos a la segunda parte del proyecto, enfocándonos en aplicar varios métodos de clasificación supervisada para determinar si los tumores de mama son benignos o malignos. Primero, empleamos el modelo de K Vecinos más Cercanos (KNN), el cual “se trata de un popular modelo supervisado que se utiliza tanto para la clasificación como para la regresión, y resulta útil para comprender las funciones de distancia, los sistemas de votación y la optimización de hiperparámetros” (Shafi, 2024, párr. 1). Comenzamos estandarizando los datos y calculando la cantidad y proporción de tumores benignos y malignos. Luego, dividimos los datos en conjuntos de entrenamiento y prueba utilizando la función “createFolds”. Ajustamos el modelo y realizamos predicciones con los datos de entrenamiento y prueba, evaluando la tasa de aciertos para medir su rendimiento. Para crear reglas de clasificación interpretables, utilizamos Árboles de Decisión. Dividimos los datos en entrenamiento (80%) y prueba (20%) y ajustamos el modelo con la función “rpart”. Posteriormente, graficamos el árbol de decisión resultante y calculamos las predicciones tanto para los datos de entrenamiento como para los de prueba, evaluando el modelo en ambos conjuntos.
Para capturar relaciones complejas y no lineales, utilizamos Redes Neuronales Artificiales (ANN), los cuales “son modelos computacionales inspirados en las neuronas biológicas, y que están conformadas por un conjunto de unidades de cómputo básico (neuronas) las cuales están conectadas entre ellas de múltiples maneras. Estas conexiones estarán definidas por unos pesos los cuales determinarán la fuerza o importancia de dichas conexiones, y durante el proceso de aprendizaje o entrenamiento de la red, serán estos pesos los que se ajustarán con el fin de producir la salida adecuada según la entrada que se aplique a la red” (Delgado, 2018). Construimos el modelo de red neuronal con los datos de entrenamiento utilizando la función “neuralnet” y graficamos la red resultante. Evaluamos el modelo generando predicciones y calculando las tasas de aciertos tanto en los datos de entrenamiento como en los de prueba. Finalmente, empleamos el método de “Máquinas de Vectores de Soporte” (SVM). “En este algoritmo, cada elemento de datos se traza como un punto en un espacio de n dimensiones (donde n es un número de características), siendo el valor de cada característica el valor de una coordenada particular. Luego, la clasificación se realiza encontrando el hiperplano que mejor diferencia las dos clases (GeeksforGeeks, 2023). Para ejecutar esta metodología utilizamos las funciones “svm” y “tune” de la librería “e1071”. “La función “svm” “se utiliza para entrenar una máquina de soporte vectorial. La función “tune” “ajusta los hiperparámetros de métodos estadísticos utilizando una búsqueda en cuadrícula sobre los rangos de parámetros proporcionados” (Meyer et al., 2022, pp. 52 y 570).
Ajustamos el modelo utilizando la función “svm”, determinando el valor de penalización óptimo para minimizar el error. Con la función “tune”, encontramos el mejor modelo, realizamos predicciones y calculamos las tasas de aciertos en los conjuntos de datos de entrenamiento y prueba.
Pasamos a la tercera parte de nuestro proyecto de investigación, enfocándonos en crear clasificaciones mediante métodos de clasificación no supervisada. En esta fase, utilizamos las técnicas “Clustering Jerárquico” y “Métodos de Particionamiento” para explorar y descubrir estructuras ocultas en los datos sin utilizar las etiquetas del diagnóstico ya establecido en la base de datos. Primero, aplicamos el “Clustering Jerárquico”, método el cual agrupa muestras en una jerarquía que se visualiza mediante un dendograma, revelando similitudes entre los tumores (Alonso, 2021). Comenzamos estandarizando los datos para asegurar que todas las características contribuyeran de manera equitativa al análisis. Luego, calculamos la matriz de distancias y creamos una visualización de esta. Utilizamos la función “fviz_nbclust”, de la librería factoextra, para determinar el número óptimo de clústeres mediante diversas metodologías, tales como gráficos de curvas como “‘silhouette’ o wss” (Datanovia, n.d). Una vez determinado este valor, ajustamos el modelo y generamos dendogramas en diferentes formatos (regular, filogenético y circular) para visualizar las clasificaciones y relaciones entre los grupos de tumores. Por último, evaluamos la cantidad de elementos por grupo según el modelo creado, analizando las clasificaciones creadas y su significado.
Luego, implementamos el Método de Particionamiento, específicamente K-media. Este método divide los datos en un número predefinido de clústeres basándose en la proximidad a centroides, ayudando a identificar subgrupos homogéneos de tumores (Alonso, 2021). Comenzamos verificando el número óptimo de clústeres (k) utilizando las funciones “NbClust” y “clValid”. Tras esto, escalamos la base de datos para asegurar uniformidad en las características, y luego calculamos la matriz de distancias. Generamos un dendograma para visualizar las clasificaciones tanto en formato regular como filogenético. Finalmente, de la misma manera que lo realizamos en el método previo, evaluamos la cantidad de elementos por grupo según el modelo, analizando las clasificaciones creadas y su significado.
Estos métodos nos permitieron detectar patrones y subtipos de tumores, mejorando nuestra comprensión de la enfermedad y potencialmente guiando nuevas investigaciones y tratamientos. Al identificar subgrupos homogéneos y similitudes entre los tumores, obtuvimos una visión más detallada de la estructura interna de los datos, lo cual es crucial para desarrollar estrategias de diagnóstico y tratamiento más precisas.
Resultados
Tras realizar los procedimientos pertinentes y crear los modelos de clasificación bajo los diferentes métodos previamente mencionados y discutidos, obtuvimos los siguientes resultados. En cuanto a la limpieza y estructuración de los datos, se encontró que la base de datos no tiene duplicados y datos faltantes, lo que es muy bueno para la investigación. El Gráfico 1, generado por la función “ggpairs”, muestra en una matriz la relación entre múltiples variables. Cada celda incluye gráficos de dispersión o coeficientes de correlación para facilitar la visualización de patrones entre pares de variables. Se pudo identificar fuertes correlaciones positivas entre las variables referentes a la media del área, perímetro y radio de los tumores. Esto es lógico porque estas tres medidas están estrechamente relacionadas con la forma y tamaño de los tumores. Por ejemplo, es esperable que un tumor con un radio grande tenga también un área y perímetro significativos. Esta correlación positiva puede indicar coherencia y consistencia en las mediciones, lo cual es valioso para el análisis. También se identificaron correlaciones positivas significativas para las variables concavidad y puntos cóncavos, respecto al resto de las variables. También se encontró que los datos en su mayoría están ligeramente sesgados a la derecha. Por último, determinamos que la cantidad de tumores benignos en la base de datos era de 357 y malignos 212, haciendo una distribución de 63% para tumores benignos y 37% para tumores malignos.

Gráfico 1: Matriz de relación entre variables, 2024
Fuente: Elaboración nuestra a partir de los datos del repositorio de aprendizaje automático de la UC Irvine
Para el método de clasificación supervisada de k vecinos más cercanos, dividimos nuestros datos en dos conjuntos: uno de entrenamiento con 455 observaciones y otro de prueba con 114 observaciones. Estos datos fueron estandarizados para un análisis coherente. Después de ajustar el modelo, encontramos que el número óptimo de vecinos (k) es 10. En los datos de entrenamiento, logramos predecir correctamente 283 de 285 tumores benignos y 165 de 170 tumores malignos, lo que representa una tasa de aciertos del 98%. En los datos de prueba, acertamos 72 de 72 tumores benignos y 39 de 42 tumores malignos, con una tasa de aciertos del 97%.
Para el método “Arboles de decisión” dividimos nuestra base de datos en entrenamiento y prueba con un total de 455 observaciones para entrenamiento y 114 observaciones para prueba utilizando otra metodología. Tras analizar los resultados, identificamos que solo se requirieron tres pasos para clasificar los diagnósticos de los tumores. Las variables que mostraron mayor influencia en esta clasificación fueron “concave_points_mean”, “area_worst”, “concavity_worst” y “perimeter_worst”. Estos resultados sugieren que estas características juegan un papel fundamental en la predicción del tipo de tumor, lo que destaca su importancia en el proceso de diagnóstico y clasificación de tumores mamarios. Tras hacer las predicciones con los datos de entrenamiento, notamos que las predicciones calculadas para los datos de entrenamiento son un 96.7% acertadas, lo que es muy bueno, siendo acertados 277 de los 282 tumores benignos y 163 de los 173 tumores malignos. Para las predicciones con los datos de prueba, notamos que las predicciones calculadas para los datos de entrenamiento son un 91.2% acertadas, siendo 70 de los 75 tumores benignos y 34 de los 39 tumores malignos acertados. Aunque este porcentaje sigue siendo positivo, muestra que el modelo puede mejorar en la generalización de la clasificación a nuevos datos.
En cuanto al método de "Redes Neuronales Artificiales", tras ajustar el modelo, calculamos las predicciones con los datos de entrenamiento previamente creados y obtuvimos que se logró acertar 282 de los 282 tumores benignos en los datos de entrenamiento y 172 de los 173 tumores malignos en los datos de entrenamiento, reflejando así una tasa de aciertos para el modelo del 99.8%, lo que es excelente y altamente preciso. Para los datos de prueba, se logró acertar 75 de los 75 tumores benignos y 36 de los 39 tumores malignos en los datos de prueba, reflejando así una tasa de aciertos para el modelo del 97% lo que es excelente. Este modelo demostró ser el mejor modelo utilizando metodología supervisada, teniendo la tasa de aciertos más alta entre los modelos creados.
Para culminar con la clasificación supervisada, pasamos al método “Maquina de Vector Soporte”. Después de ajustar el modelo y determinar el valor de penalización óptimo, logramos una tasa de aciertos del 98% en los datos de entrenamiento. Esto se tradujo en la correcta identificación de 280 de 282 tumores benignos y 169 de 173 tumores malignos. Las predicciones con los datos de prueba mantuvieron esta alta precisión, con una tasa de aciertos del 98%, clasificando correctamente 75 de 75 tumores benignos y 36 de 39 tumores malignos. Este modelo resalta como otro enfoque altamente efectivo para la clasificación de tumores mamarios.
Pasando a analizar los resultados de la metodología de clasificación no supervisada, empleamos dos enfoques: Clustering Jerárquico y Métodos de Particionamiento. En el caso del Clustering Jerárquico, se determinaron números óptimos de clústeres mediante los métodos "silhouette", "wss" y "gap_stat" (mostrados en Gráfico 2, Gráfico 3 y Gráfico 4), donde Silhouette mide qué tan bien se agrupan los puntos, con valores más altos indicando mejor cohesión. WSS evalúa la suma de distancias cuadradas a los centroides, y el "codo" en su gráfica sugiere el número ideal de clústeres. Finalmente, el “gap_stat” ayuda a encontrar el número óptimo de clústeres al comparar la variabilidad interna de los clústeres con datos aleatorios, eligiendo el valor donde el gap es mayor o se estabiliza. Estas metodologías fueron aplicadas utilizando la librería “factoextra”, la cual “es un paquete de R que facilita la extracción y visualización de los resultados de análisis exploratorios de datos multivariados” (Kassambara & Mundt, 2022). Resultando en 2, 4 y 2 clústeres respectivamente, tras analizar las gráficas realizadas. Optamos por utilizar 4 clústeres para visualizar los resultados ya que, aunque ya sabemos que nuestro objetivo es clasificar los datos en dos grupos (benignos y malignos), al trabajar con solo dos clústeres, se observaba una distribución desigual, en la que un grupo aparecía mucho más grande que el otro (aproximadamente 98% frente a 2%). Sabemos que esta visualización no es precisa, ya que previamente calculamos que la distribución real es de 63% de tumores benignos y 37% malignos para la base de datos original. Este error gráfico puede deberse a varios factores, como el uso de datos estandarizados y las particularidades del método de clustering. Sin embargo, al utilizar cuatro clústeres, los grupos benignos y malignos se distinguen con mayor claridad y, además, se observan datos que se diferencian lo suficiente como para que los modelos no los clasifiquen en ninguno de estos dos grupos. Esto sugiere un pequeño margen de error en los modelos, el cual podría corresponder a casos atípicos o muestras con características únicas. Obtuvimos las siguientes clasificaciones: 177, 7, 383 y 2 elementos para los grupos 1, 2, 3 y 4, respectivamente. En cuanto al método de "Métodos de Particionamiento", se estableció que los números óptimos de clústeres eran 4 y 2 según dos métodos diferentes. Decidimos seguir con la elección de 4 clústeres como en el método anterior ya que observamos el mismo problema de distribución desigual que en el método anterior, donde un grupo visualmente predominaba sobre el otro cuando usábamos solo dos clústeres. Además, queremos mantener una metodología consistente entre ambos métodos de clasificación no supervisada, asegurando que los resultados sean comparables y que las visualizaciones reflejen más claramente las posibles variaciones entre los datos benignos y malignos. Los resultados mostraron una distribución de elementos por grupo similar a la obtenida previamente, con 177, 7, 383 y 2 elementos para los grupos 1, 2, 3 y 4, respectivamente.
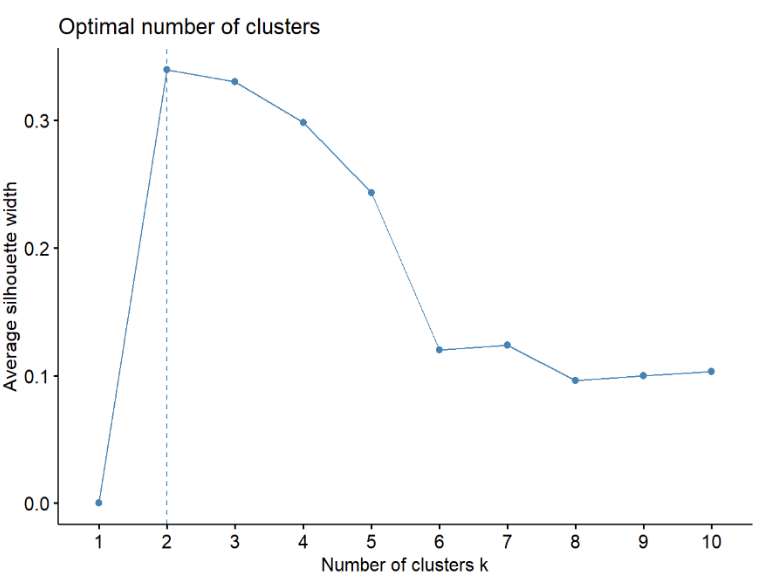
Gráfico 2: Número óptimo de clústeres utilizando método “Silhouette”, 2024
Fuente: Elaboración nuestra a partir de los datos del repositorio de aprendizaje automático de la UC Irvine

Gráfico 3: Número óptimo de clústeres utilizando método “WSS”, 2024
Fuente: Elaboración nuestra a partir de los datos del repositorio de aprendizaje automático de la UC Irvine

Gráfico 4: Número óptimo de clústeres utilizando método “Gap Stat”, 2024
Fuente: Elaboración nuestra a partir de los datos del repositorio de aprendizaje automático de la UC Irvine
El análisis de los resultados obtenidos mediante la metodología de clasificación no supervisada revela la eficacia de dos enfoques distintos: Clustering Jerárquico y Métodos de Particionamiento. Estos métodos permiten agrupar los datos de manera automática, identificando patrones y estructuras subyacentes sin la necesidad de etiquetas predefinidas. En nuestro estudio, aplicamos estos enfoques para comprender la distribución de tumores mamarios en diferentes grupos con características similares. Al evaluar el Clustering Jerárquico, empleamos métodos de evaluación de clústeres como "silhouette", "wss" y "gap_stat" para determinar el número óptimo de clústeres.
Conclusiones
La combinación de diferentes métodos de clasificación supervisada ha arrojado resultados sobresalientes en la predicción de tumores mamarios benignos y malignos en pacientes con cáncer de mama. El método de k vecinos más cercanos demostró un rendimiento sobresaliente, con tasas de aciertos del 98% en los datos de entrenamiento y del 97% en los datos de prueba. Esto resalta su alta capacidad para distinguir entre ambos tipos de tumores, lo que puede tener un impacto significativo en la precisión diagnóstica y, por ende, en la atención y el tratamiento de los pacientes. Los Árboles de Decisión mostraron una efectividad notable al clasificar los diagnósticos de tumores en tan solo tres pasos. Identificar las variables clave como “concave_points_mean”, “area_worst”, “concavity_worst” y “perimeter_worst” resalta su importancia en este proceso. Aunque la tasa de aciertos en los datos de prueba fue ligeramente menor, el modelo aún demuestra una sólida capacidad de clasificación. Las Redes Neuronales Artificiales destacaron con una tasa de aciertos del 99.8% en los datos de entrenamiento y del 97% en los de prueba, demostrando una capacidad excepcional para la clasificación precisa de tumores. Su alto rendimiento respalda su utilidad en entornos clínicos y mejora de diagnósticos relacionados con el cáncer de mama.
Finalmente, la Máquina de Vectores de Soporte (SVM) demostró ser altamente efectiva, con tasas de aciertos del 98% tanto en entrenamiento como en prueba. Estos resultados subrayan la eficacia y confiabilidad de estas técnicas en la mejora de la precisión y eficacia de los diagnósticos en el cáncer de mama, lo que puede traducirse en mejores resultados médicos para los pacientes. La aplicación de métodos de aprendizaje automático supervisado es fundamental para avanzar en la precisión de los diagnósticos en esta área crucial de la salud.
Esta selección meticulosa nos llevó a optar por utilizar 2 grupos para visualizar los resultados. En contraste, el Método de Particionamiento nos sugirió 4 clústeres como óptimos según dos enfoques diferentes. Aunque existieron diferencias en la determinación del número de clústeres, la distribución de elementos por grupo se mantuvo consistente entre ambos enfoques, destacando la robustez de los resultados obtenidos. La elección de la cantidad adecuada de clústeres es crucial para una interpretación significativa de los datos. En nuestro caso, esta decisión se basó en la representación adecuada de los patrones encontrados en los tumores mamarios. La consistencia en la distribución de elementos por grupo en ambos métodos subraya la relevancia y confiabilidad de los patrones identificados.
En términos prácticos, estos resultados tienen importantes implicaciones en la segmentación y comprensión de conjuntos de datos complejos como los relacionados con el cáncer de mama. Tanto Clustering Jerárquico como Métodos de Particionamiento ofrecen herramientas valiosas para explorar y entender la estructura subyacente de los datos, lo que puede ser fundamental para la toma de decisiones clínicas y la investigación médica.
Referencias
Alonso, C. (2021). Clustering: K-means. ETSI. https://dcain.etsin.upm.es/~carlos/bookAA/03.1_Clustering-K-Means.html
Datanovia. (n.d.). Determining the optimal number of clusters: 3 must-know methods. https://www.datanovia.com/en/lessons/determining-the-optimalnumber-of-clusters-3-must-know-methods/
Delgado, R. (2018). Determining the optimal number of clusters: 3 must-know methods. RPubs. https://rpubs.com/rdelgado/402754
GeeksforGeeks. (2023). Classifying data using support vector machines (SVMs) in R. https://www.geeksforgeeks.org/classifying-data-using-supportvector-machinessvms-in-r/
Kassambara, A., & Mundt, F. (2022). Extract and Visualize the Results of Multivariate Data Analyses. http://www.sthda.com/english/rpkgs/factoextra
Meyer, D., Dimitriadou, E., Hornik, K., Weingessel, A., & Leisch, F. (2022). e1071: Misc Functions of the Department of Statistics, Probability Theory Group (Formerly: E1071), TU Wien. https://cran.r-project.org/web/packages/e1071/e1071.pdf
Schloerke, B. (2015, October 29). ggpairs: A pairwise plot function for GGobi. https://ggobi.github.io/ggally/articles/ggpairs.html
Shafi, A. (2024). K-nearest neighbor classification with scikit-learn. DataCamp. https://www.datacamp.com/es/tutorial/k-nearest-neighbor-classification-scikitlearn
Waring, E. (2022). skimr: Compact and flexible summaries of data (Version 2.1.5). https://www.rdocumentation.org/packages/skimr/versions/2.1.5
Wolberg, W., Mangasarian, O., Street, N., & Street, W. (1995). Breast ancer Wisconsin (Diagnostic). UCI Machine Learning Repository. https://doi.org/10.24432/C5DW2B
Esta obra está bajo una licencia de Creative Commons Reconocimiento-NoComercial 4.0 Internacional.